Modifications
General Approach
Most protein samples exhibit some degree of modification.
There are the "natural" post translational modifications, such as phosphorylation and glycosylation. There are the accidental modifications which are artefacts of sample handling, such as oxidation. Finally, there are the modifications deliberately introduced during sample work-up, such as cysteine derivatisation. In most cases, it is only the deliberate modifications which are known about for certain at the time of doing a search.
It might be assumed that the search software could allow for those modifications which are described in sequence entry annotations. However, writing code to parse these sequence annotations would be a major task. Indeed, many post-translational modifications are not specified in a way which can be readily translated into specific mass differences. For example, noting that a residue is an actual or potential glycosylation site is not much help. Even a simple modification, such as phosphorylation, is rarely quantitative, so that it would be necessary to include mass values for all permutations of occupied and unoccupied sites.
And, of course, protein sequences derived translated from nucleotide sequences contain no information on post translational modifications.
The solution adopted here is to allow modifications to be specified in two different ways: fixed modifications and variable modifications. (Quantitation methods support an additional mode: Exclusive modifications.)
Fixed modifications are applied universally, to every instance of the specified residue or terminus. There is no computational overhead associated with a fixed modification, it is simply equivalent to using a different mass for the modified residue or terminus. For example, selecting Carboxymethyl (C) as a fixed modification means that all calculations will use 161 Da as the mass of cysteine.
Variable modifications
Variable modifications are those which may or may not be present. Mascot tests all possible arrangements of variable modifications to find the best match. For example, if Oxidation (M) is selected as a variable modification, and a peptide contains 3 methionines, Mascot will test for a match with the experimental data for that peptide containing 0, 1, 2, or 3 oxidised methionine residues. This greatly increases the complexity of a search, resulting in longer search times and reduced specificity, so variable modifications should be used sparingly.
If a search contains a variable modification with the same specificity as a fixed modification, the variable can displace the fixed. For example, if Carbamidomethyl (C) and Propionamide (C) were both variable modifications, there would be three possibilities each Cysteine: modified with Carbamidomethyl, modified with Propionamide, or unmodified. If Carbamidomethyl (C) was fixed and Propionamide (C) was variable, then you would only see one or the other; you would never see an unmodified Cysteine.
When a search contains many variable modifications, there may be a large number of possible modification states for a candidate peptide that agree with the experimental mass. It is necessary to place an upper limit on the number of possibilities that are tested and scored, otherwise searches would become unacceptably slow. To illustrate how these limits work, consider an example:
Variable modifications selected for the search: Acetyl (K), Acetyl (N-term), Methyl (K), Dimethyl (K), Trimethyl (K)
Candidate peptide: QLATKAARKSAPSTGGVKKPHRYKPGTVALK with m/z corresponding to a modification delta of 126 Da
Assume that the mass accuracy is +/- 0.2 Da, so that we cannot tell whether a peptide is modified by Acetyl (42.01 Da) or Trimethyl (42.05 Da)
Limits set in mascot.dat:
- A limit on the number of distinct varmods found on a single peptide: MaxPepNumVarMods=3
(3 x Acetyl (K) is a single distinct varmod, 1 x Acetyl (N-term) + 2 x Acetyl (K) is two distinct varmods)- A limit on the number of modified sites found on a single peptide: MaxPepNumModifiedSites=5
- A limit on the number of arrangements of an individual varmod composition: MaxPepModArrangements=64
The first step is to enumerate all possible varmod compositions that fit to the experimental precursor mass of the candidate peptide. The constraints on this list are MaxPepNumVarMods, MaxPepNumModifiedSites, and the fact that the candidate peptide contains six K residues and one N-term. For example, the total delta of 126 could be any of the following compositions:
- 3 x Acetyl (K)
- 3 x Trimethyl (K)
- 1 x Acetyl (N-term), 2 x Acetyl (K)
- 1 x Acetyl (N-term), 1 x Acetyl (K), 1 x Trimethyl (K)
- 1 x Acetyl (N-term), 2 x Trimethyl (K)
- 2 x Acetyl (K), 3 x Methyl (K)
- and many others
But, it could not be one of these compositions, even though they add up to the correct delta mass:
- 2 x Acetyl (N-term), 1 x Acetyl (K) – only one N-term available
- 3 x Methyl (K), 3 x Dimethyl (K) – exceeds MaxPepNumModifiedSites
- 1 x Acetyl (K), 1 x Methyl (K), 1 x Dimethyl (K), 1 x Trimethyl (K) – exceeds MaxPepNumVarMods
For each composition that fits the required delta mass, multiple arrangements may be possible. Different arrangements have the same peptide mass but will give rise to differences in the MS/MS spectrum. For example, there are 20 possible arrangements of 3 x Acetyl on 6 x K, here shown schematically:
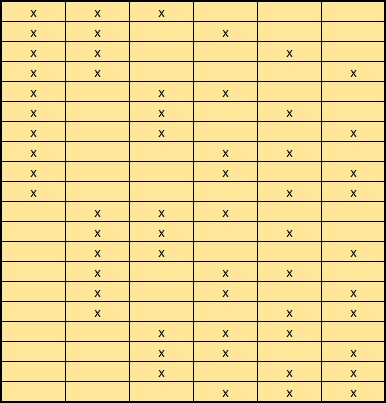
If the number of arrangements of an individual composition is less than MaxPepModArrangements, all can be tested and the highest scoring match reported. If the number of arrangements is greater than MaxPepModArrangements, arrangements are tested in random order, so that the entire space of possible arrangements is sampled before reaching the limit.
For example, there are 180 possible arrangements of 1 x Acetyl (K) + 2 x Methyl (K) + 2 x Dimethyl (K), of which only 64 would be tested. Even if this happened to be the correct composition, there is only a 1 in 3 chance that the perfectly correct arrangement will be one of those that are scored. Even if the MS/MS spectrum is of very high quality, the reported match is likely to be a nearly correct arrangement rather than the perfectly correct arrangement. If finding the best possible match is important, and this was a possible composition, you would need to increase MaxPepModArrangements and accept that the search would be slower.
Whatever the limits, for best speed and specificity, it is essential to minimise the number of variable modifications included in a search. For example, if the mass accuracy does not allow Acetyl (K) and Trimethyl (K) to be distinguished, one should be dropped. If the interest is in post translational modifications, select Acetyl (Protein N-term), not Acetyl (N-term).
Unimod
The list of modifications used by Mascot is taken directly from the Unimod database. For further details of individual modifications, please refer to Unimod. Note that Unimod is a community supported resource. If you want to add a new modification to Unimod, you can do so, and you then become the curator of the new record. The Mascot modifications list on the public web site is updated from Unimod each weekend.
By default, only selected modifications are displayed in the Mascot search form. If you want to see the complete list, you must go to the search form defaults page and tick the checkbox for ‘Show all mods.’.
In Mascot 2.1 and earlier, modification definitions were stored in a configuration file called mod_file. Mascot now takes its modification definitions direct from an XML representation of the Unimod database. To update the local definitions, simply download the latest XML file from the Unimod help page.
In Unimod, both amino acid residues and modifications are defined in terms of their elemental composition. This is important for metabolic labelling, in which the isotopic label is present throughout the peptide backbone. If you want to view or edit the local unimod.xml file, a browser-based Configuration Editor is provided:
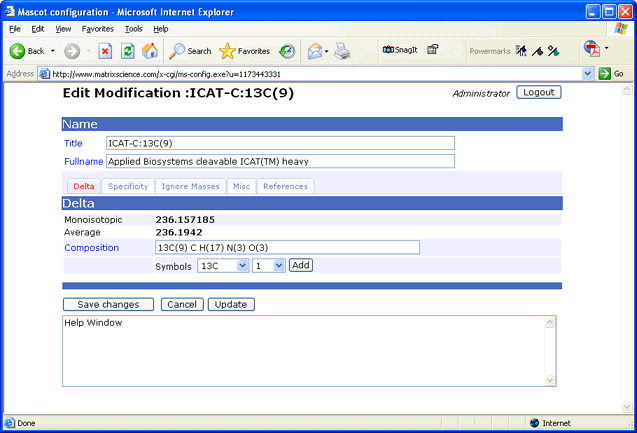
Note: Whenever unimod.xml is updated, an equivalent mod_file is created automatically to support old client applications that require this file. Do not be tempted to edit mod_file, because any changes will be lost the next time unimod.xml is updated.
Other lists of modifications
DeltaMass is a comprehensive list of modifications, sorted by mass.
RESID database contains detailed descriptions of many post-translational modifications.
Neutral Losses
Unimod supports four types of neutral loss
Scoring: A neutral loss from the MS/MS fragments. The resultant ions are considered for scoring, e.g. y-98 or b-98 for phosphopeptides. There can be up to 10 scoring neutral losses. During a search, if there are multiple neutral losses, Mascot iterates through the scoring ones. The loss that gives the highest score is chosen, and all the other neutral losses are treated as Satellite.
Satellite: A neutral loss specified as satellite is never considered for scoring. If a Satellite neutral loss gives a match to a peak, that peak is removed from the list of noise peaks, which improves the score. None of the standard modifications in Unimod currently have satellite neutral losses.
Peptide: A neutral loss from the intact peptide precursor. This peak is matched and so not treated as a noise peak for scoring purposes
Required Peptide: A required peptide neutral loss must be present in the spectrum. This carries some risk, because a perfectly good match could be rejected if this peak was missing.
Phosphorylation
Phosphorylation is one of the most interesting and studied modifications. It is also one of the most challenging for database searching, because of these factors:
- Site heterogeneity
- 3 fragmentation channels
- intact fragments
- neutral loss of HPO3 (80 Da)
- neutral loss of H3PO4 (98 Da)
- Can occur at STY – ~16% of residues.
Support for a single neutral loss per modification was introduced in Mascot 1.7. Mascot 2.1 added support for multiple neutral losses from both fragment ions and the precursor.
In the default phosphorylation modifications derived from Unimod, pY fragments always stay intact, while pS and pT fragments can stay intact or can lose 98.
This is not a hard and fast rule, and sometimes a loss of 80 is also observed. However, this is not included in the definition because it is identical to the delta of the original modification. Allowing for the possibility of 80 Da neutral loss introduces ambiguity as to the site of the modification when there are multiple potential phosphorylation sites in a peptide. For example, this match to pTESPATAAETASEELDNR gets a score of 115
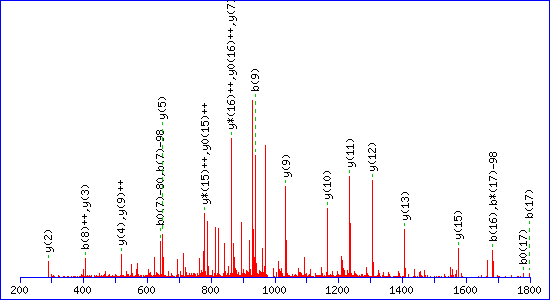
If a neutral loss of 80 Da is allowed, the score for a match to TESPATAAETApSEELDNR is almost as high, 92
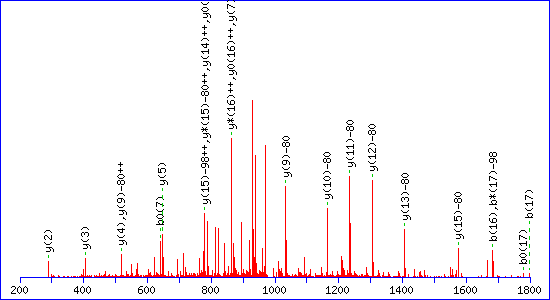
The reason is clear. The matching peaks are all y ions, so the point of modification can be shifted towards the C-terminus by swapping the matching series from y to y-80. Without the availability of an 80 Da loss, the score for the second match drops to 29.
It has often been observed that the neutral loss from the precursor can be an excellent guide to the identity of the phosphorylated residue. If a strong loss of 98 Da is observed, then the expectation is pS or pT. If no neutral loss, then pY. In Mascot, one or more precursor neutral losses can be specified. They can also be made "required", which means that the peak must be present in the spectrum. This carries some risk, because a perfectly good match could be rejected if this peak happened to be missing.
Site Analysis
If a peptide has two serines and a single phosphate on one of them, there may or may not be evidence in the MS/MS spectrum to favour one site over the other. It depends on the separation of the two sites, whether there are sequence ions in the region between the potential sites, and the signal to noise for the assignable fragment ion peaks. If the result report shows matches to both possibilities, our rule of thumb used to be that a score difference 20 or more meant that the lower scoring match could be neglected. See, for example, Phosphorylation – how reliable is site analysis?
This concept has since been quantified by Bernard Kuster’s group at the Technische Universitaet Muenchen into the Mascot Delta Score or MD-score. This is described in detail in Savitski, M. M., et al. (2011). "Confident Phosphorylation Site Localization Using the Mascot Delta Score." MCP 10: M110.003830. Very briefly, a collection of 180 synthetic analogs of natural phosphopeptides was analysed to quantify the accuracy of using the score difference between the top two matches. This made it possible to determine the false localisation rate for a given score difference. As might be expected, the numbers were observed to have some dependency on instrument characteristics and ionisation method.
The default setting in Mascot is slightly more conservative than the FLR data reported by Kuster, such that two matches with an MD-score of 10 will be reported as ‘probabilities’ of 91% and 9%. This is based on the Mascot score being -10LogP, where P is the probability of the match being random. Hence, a difference of 10 in the score corresponds to a factor of 10 in the probability of the peptide sequence match. The sensitivity can be adjusted using a global parameter setting in the options section of mascot.dat. The default corresponds to SiteAnalysisMD10Prob 0.1. Decrease this value (e.g. to 0.05) to make the numbers more conservative. If you are tempted to increase the setting (e.g. to 0.2) to make the effect for a given score difference more dramatic, we recommend testing the accuracy of the results by analysing some known standards, as in Kuster’s work.
Site analysis is performed whenever the top rank match is significant and contains one or more variable modifications for which alternative arrangements are possible. The results are displayed in the Peptide View report. For example, using the default setting produces the following results:
| Score | Mr(calc) | Delta | Sequence | Site Analysis |
|---|---|---|---|---|
| 83.4 | 1846.7179 | 0.1889 | DIGSESTEDQAMEDIK | Phospho S4 84.56% |
| 75.8 | 1846.7179 | 0.1889 | DIGSESTEDQAMEDIK | Phospho S6 14.73% |
| 62.7 | 1846.7179 | 0.1889 | DIGSESTEDQAMEDIK | Phospho T7 0.72% |
| 26.9 | 1846.7808 | 0.1261 | KLNSNPENYCESELK | |
| 22.8 | 1846.7729 | 0.1339 | KMEDSVGCLETAEEVK | |
| 15.5 | 1846.9230 | -0.0161 | GAYTIEQHPVLGLEIK | |
| 14.2 | 1846.7729 | 0.1339 | KMEDSVGCLETAEEVK | |
| 13.9 | 1846.8754 | 0.0315 | YVKGIYENLPSIDEK | |
| 13.8 | 1846.8866 | 0.0202 | QLIEAPDPVPSFEVAR | |
| 13.3 | 1846.9052 | 0.0016 | KIDFSNIAMLFGGVQK |
A large score difference will strongly favour one arrangement
| Score | Mr(calc) | Delta | Sequence | Site Analysis |
|---|---|---|---|---|
| 84.5 | 3541.7900 | 0.0191 | KRYGASAGNVGDEGGVAPNIQTAEEALDLIVDAIK | Deamidated N9 99.79% |
| 57.2 | 3541.7900 | 0.0191 | KRYGASAGNVGDEGGVAPNIQTAEEALDLIVDAIK | Deamidated N19 0.19% |
| 47.9 | 3541.7900 | 0.0191 | KRYGASAGNVGDEGGVAPNIQTAEEALDLIVDAIK | Deamidated Q21 0.02% |
| 14.3 | 3541.7735 | 0.0355 | INKRLNYIKRQPHQSDDEPAQIMGYKNK | |
| 14.3 | 3541.7735 | 0.0355 | INKRLNYIKRQPHQSDDEPAQIMGYKNK | |
| 13.5 | 3541.7470 | 0.0620 | ENEVPERKNYEDEMQVTKLPVNQNILKN | |
| 13.0 | 3541.8013 | 0.0078 | RNVISQINDGQVQVTTQKLPHPVSQIGDGQIQ | |
| 12.9 | 3541.7472 | 0.0618 | ALLVMSDKVYENYTNNINFYMSKNLIKK | |
| 12.8 | 3541.8641 | -0.0551 | IRSTFKYSPINNPNLILDVKNGSGNEQRPTI | |
| 12.6 | 3541.7472 | 0.0618 | ALLVMSDKVYENYTNNINFYMSKNLIKK |
When there is little to choose between two arrangements, this could indicate a lack of evidence or it could indicate a mixture of the two forms. There is nothing in the algorithm to distinguish between these possibilities.
| Score | Mr(calc) | Delta | Sequence | Site Analysis |
|---|---|---|---|---|
| 73.1 | 4178.0808 | 0.0369 | KIATYQERDPANLPWGSSNVDIAIDSTGVFKELDTAQK | Deamidated N19 42.20% |
| 72.5 | 4178.0808 | 0.0369 | KIATYQERDPANLPWGSSNVDIAIDSTGVFKELDTAQK | Deamidated N12 37.01% |
| 70.0 | 4178.0808 | 0.0369 | KIATYQERDPANLPWGSSNVDIAIDSTGVFKELDTAQK | Deamidated Q6 20.72% |
| 45.4 | 4178.0808 | 0.0369 | KIATYQERDPANLPWGSSNVDIAIDSTGVFKELDTAQK | Deamidated Q37 0.07% |
| 21.9 | 4178.0463 | 0.0713 | ISMADNLLSTINKSEINKGFDRNLGELLLQQQQELR | |
| 15.3 | 4178.0987 | 0.0189 | TVGDYVITPDICLERKSISDLIGSLQNNRLANQCKK | |
| 15.0 | 4178.0987 | 0.0189 | TVGDYVITPDICLERKSISDLIGSLQNNRLANQCKK | |
| 15.0 | 4178.0987 | 0.0189 | TVGDYVITPDICLERKSISDLIGSLQNNRLANQCKK | |
| 15.0 | 4178.0987 | 0.0189 | TVGDYVITPDICLERKSISDLIGSLQNNRLANQCKK | |
| 15.0 | 4178.0987 | 0.0189 | TVGDYVITPDICLERKSISDLIGSLQNNRLANQCKK |

